
Brazil’s Medicines Market Regulation Chamber (CMED) has taken another significant step toward modernizing its drug pricing regulatory framework through the enactment of CM/CMED Rule No. 3/2025, a long-awaited reform that replaces the long-standing regime established under Rule No. 2/2004 and introduces a reorganised and more structured system for setting maximum entry prices for new medicines and new presentations.
The Rule responds to persistent criticism from the regulated sector that Brazil’s pricing control model had become fragmented, increasingly discretionary, and insufficiently adapted to incremental innovation and technology-intensive products. It enters into force on 29 April 2026, with the transition period intended to be used for the preparation of pricing requests for products currently pending marketing authorisation before Anvisa.
The Rule also applies to ongoing proceedings, including DIP analyses pending before CMED’s Executive Secretariat and provisional or omissive-case prices that have not yet been made definitive. Affected companies must supplement the required documentation within 30 days of the Rule’s entry into force, failing which CMED may initiate a procedure to define the initial price, with procedural deadlines restarting upon submission of the supplementary information.
This article outlines the main changes introduced by the new regulatory framework.
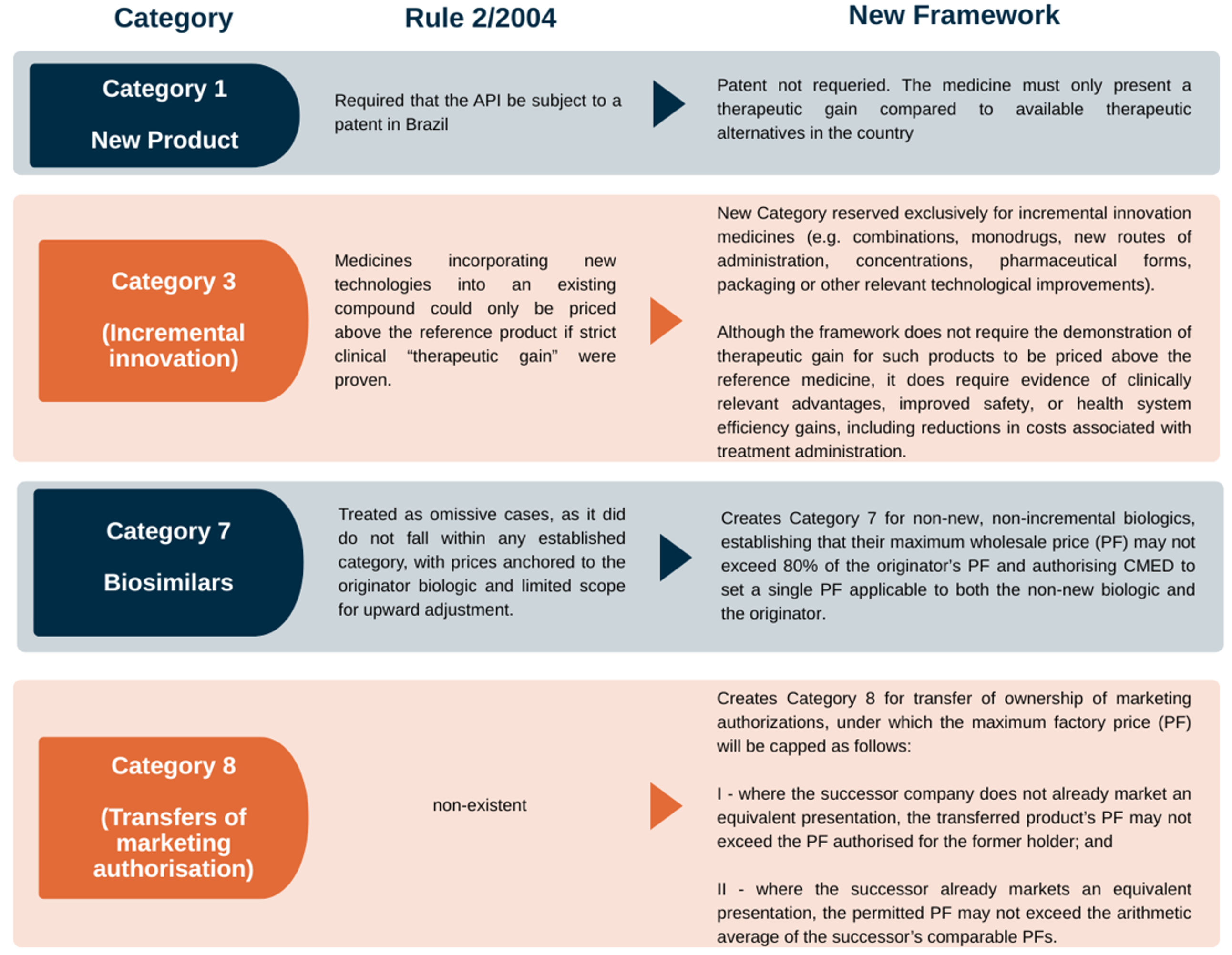
Rule No. 3/2025 establishes eight categories, which serve as the primary determinant of the applicable price ceilings and evidentiary requirements. The table below summarizes the main changes introduced to Categories 1 and 3, as well as the creation of the new Categories 7 and 8.
A particularly relevant aspect of Rule No. 3/2025 is the reformulation of the concept of therapeutic gain and, above all, the introduction of more explicit parameters for its demonstration, which has historically been one of the most contentious elements of CMED’s entry-pricing framework.
Under the former regime, the interpretation of therapeutic gain was often viewed as narrow and closely tied to strict clinical superiority criteria, which in practice limited the pricing recognition granted to products that, while not necessarily superior in efficacy endpoints, introduced meaningful improvements in safety, tolerability, convenience, adherence or treatment administration.
The new framework seeks to address this bottleneck by expressly allowing the Technical-Executive Committee to consider other scientifically supported therapeutic advantages beyond the traditional parameters (e.g., improved efficacy profile, safety gains and reduction of adverse effects), and by clarifying the types of scientific evidence that may substantiate such advantages.
Rule No. 3/2025 expands the list of reference countries from 9 to 14, including Germany, Japan, the UK, Norway, Mexico, South Africa and others, while removing New Zealand.
The international reference basket of countries plays a central role in CMED’s drug pricing regulation, as it establishes a comparative framework for defining the maximum entry price of new medicines in Brazil. At a meeting with representatives of the regulated sector held by Anvisa on July 4, 2025, the agency emphasized that updating the basket is consistent with good regulatory practices and aims to ensure greater alignment with current market conditions. The countries selected were chosen based on factors such as purchasing power for medicines, prevailing price levels, ease of access to official price information, and the speed with which new products are introduced to the market.
For instance, Argentina and China were not included due to specific challenges: in Argentina, high prices, weak regulation, and recurrent exchange rate volatility; in China, heterogeneous provincial pricing policies and language barriers. Switzerland was also not included, unlike countries such as the United States, which is a priority market for the introduction of new medicines, the high price levels in Switzerland are not justified by its limited relevance for the launch of new medicines.
Therefore, as countries with comparable purchasing power and/or pricing rules to Brazil’s are not necessarily those prioritized for the launch of new medicines, the number of countries in the basket was expanded. In addition to expanding the number of countries, the price cap for wholesale segment (PF) will be calculated based on prices market in at least four of the countries (Rule No 2/2004 provides for three). While encouraging more competitive pricing, the proposed draft requires greater regulatory strategy by the companies to avoid negative anchoring of its drug’s prices.
Where a product is not yet commercialised in at least four reference countries, CMED will establish a provisional PF and the company must thereafter submit, on an annual basis, documentation evidencing the product’s launch and respective price in the reference jurisdictions.
Rule No. 3/2025 defines a filing window for the Pricing Information Document (DIP) - the dossier containing the technical and administrative documentation required for CMED’s pricing assessment. The DIP must be submitted after filing the marketing authorisation request and before publication of its granting.
If the DIP is not submitted within this window, CMED will initiate a formal price-setting procedure and notify the responsible company to provide the required documentation within 30 days. Should the company fail to comply within such timeframe, the Executive Secretariat of CMED will define the initial PF ex officio, based solely on the information available to the authority, without prejudice to applicable legal sanctions.
The new framework further codifies the treatment of products whose marketing authorisation depends on the submission of additional evidence after approval. In such cases, CMED may establish a provisional PF until the information required for definitive pricing becomes available. The pricing decision must expressly indicate which data are essential for the definitive PF definition or provide that the final price will be set upon compliance with the commitment term executed with Anvisa.
During this period, the company must submit technical reports evidencing efficacy and safety according to the agreed schedule, under penalty of sanctions. Once the required information has been fully delivered - or once Anvisa’s commitment term has been fully complied with - CMED must define the definitive PF within 90 days. If CMED does not act within this period, the provisional PF remains valid until a final decision is issued.
Modality applicable to marketing authorisation holders whose products already have a PF established by CMED and who elect to adopt Anvisa’s simplified procedures for registration, post-registration amendments and renewals. In such cases, the company must file the DIP in simplified form, under CMED’s rules, and CMED must complete its analysis within 60 days, thereby enhancing efficiency for lifecycle management and regulatory maintenance.
Following Rule No. 2/2025, which established procedural changes in CMED’s pricing regulation 1, Rule No. 3/2025, determines mandatory review by CMED’s Technical-Executive Committee (CTE), even in the absence of an appeal by the company.
The Mandatory Review applies to: (i) decisions issued by the Executive Secretariat in omissive cases, as previously provided by Rule No. 2/2025; and (ii) decisions where the maximum PF of the Category 1 and/or 3 is defined based on the company’s proposed pricing rationale.
The main issue is that CTE may decide to lower the price already granted. Although the company may submit a motion for reconsideration with devolutive effect, the mechanism may still generate legal uncertainty and prolong the process.
Although the new framework represents a substantial milestone in the modernization of Brazil’s drug pricing regulation, Rule No. 3/2025 expressly confirms that Advanced Therapy Products (ATMPs) remain outside the core categorization and pricing criteria established for new medicines and new presentations. The Rule provides that the pricing criteria applicable to ATMPs will be established through specific act issued by CMED’s Council of Ministers, and, until such instruments are enacted, these products must be treated as omissive cases.
ATMPs have historically lacked objective and tailored pricing parameters under Rule No. 2/2004, contributing to prolonged review cycles, multiple revisions, and a higher propensity for litigation and judicialization—ultimately delaying patient access.
The persistence of this gap reflects the structural challenge of fitting ATMPs into a traditional price-cap model based on external reference pricing and conventional comparators. ATMPs exhibit distinctive characteristics that complicate entry-pricing design, including high one-time costs, customized or patient-specific manufacturing processes (e.g., cell therapies), and the need for continuous long-term efficacy assessment, often with limited real-world evidence available at the time of authorization. These features have led the regulated sector to call for a specific framework capable of capturing the real economics of ATMP development and supply while enabling risk management by public and private payers.
Following the targeted consultation conducted in 2025 on this topic, CMED has signaled an intention to advance the regulatory treatment of ATMPs, and the regulated sector broadly expects the adoption of a specific pricing framework during 2026, aimed at addressing the current gap and enhancing predictability for advanced therapies.

This section gives quick answers to the most common questions about this insight. What changed, why it matters, and the practical next steps. If your situation needs tailored advice, contact the RNA Law team.
Q1: What is the main purpose of CMED Rule No. 3/2025?
A1: CMED Rule No. 3/2025 aims to modernize Brazil's drug pricing regulatory framework, replacing a long-standing regime and introducing a more structured system for setting maximum entry prices for new medicines and presentations.
Q2: How does the new rule address the concept of "therapeutic gain"?
A2: The new framework reformulates "therapeutic gain" by allowing consideration of a wider range of scientifically supported therapeutic advantages beyond strict clinical superiority, such as improved safety, tolerability, convenience, and adherence.
Q3: What changes were made to the reference country basket for drug pricing?
A3: Rule No. 3/2025 expands the list of reference countries from 9 to 14, including new nations like Germany, Japan, and the UK, and requires pricing based on at least four reference countries instead of three.
Q4: Does Rule No. 3/2025 establish pricing criteria for Advanced Therapy Products (ATMPs)?
A4: No, Rule No. 3/2025 explicitly confirms that ATMPs remain outside its core categorization and pricing criteria, with specific guidelines expected to be established through a separate act by CMED's Council of Ministers.

.png)

